Introduction
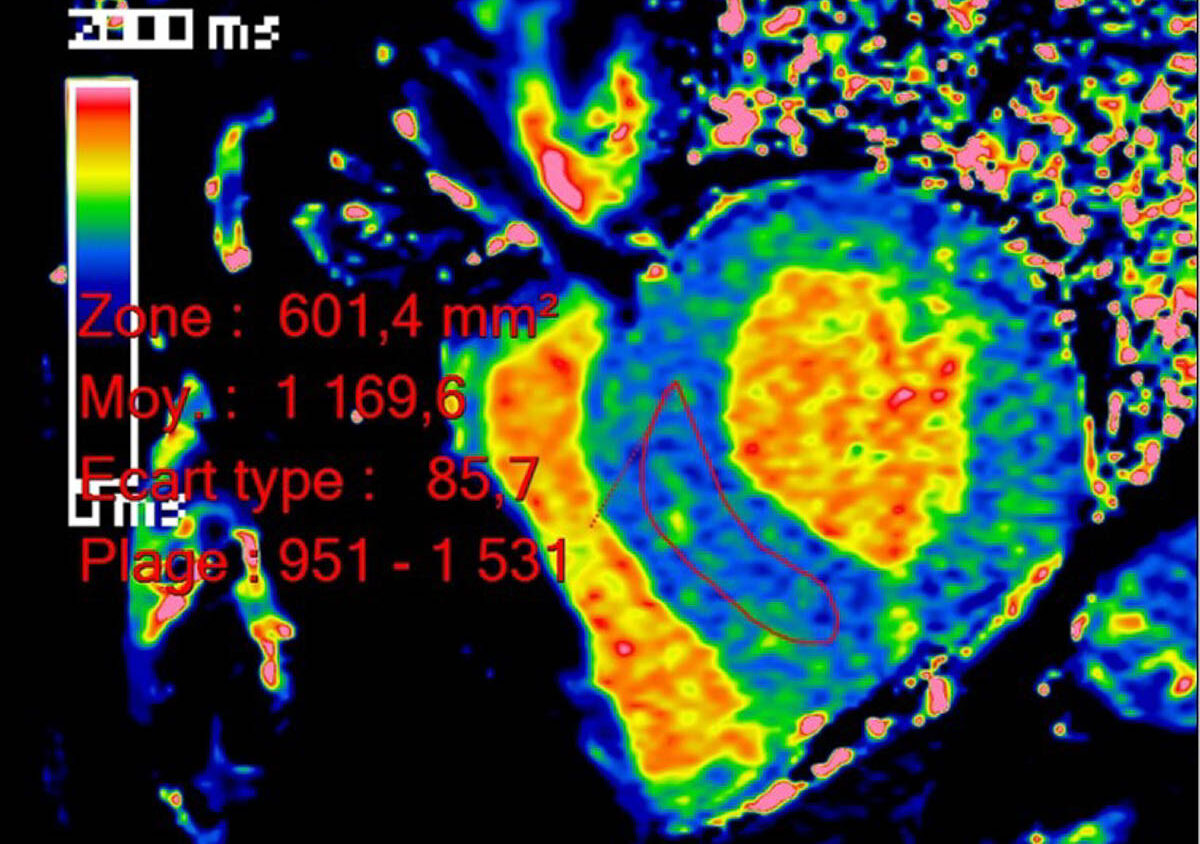
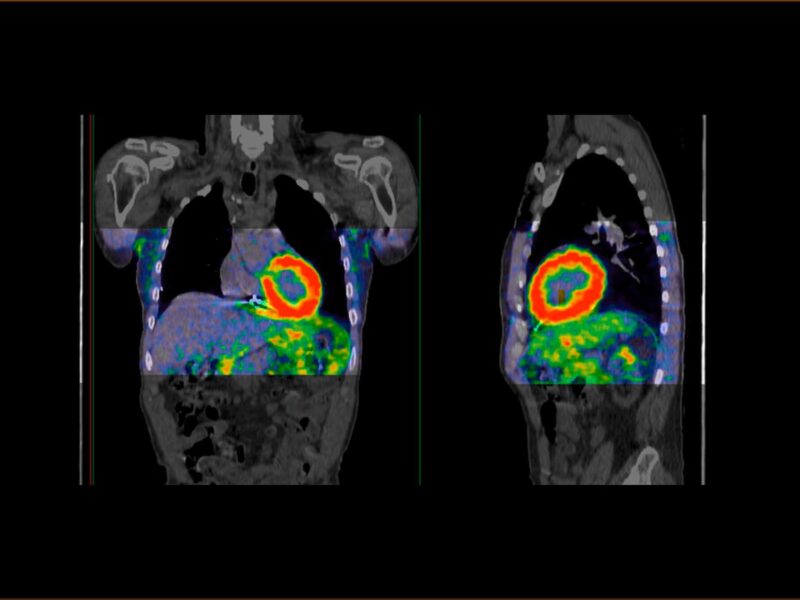
L’amylose cardiaque, ou cardiopathie amyloïde (CA), est secondaire au dépôt de fibres amyloïdes dans le cœur, entraînant une cardiopathie restrictive [1]. La fréquence de la maladie est mal connue mais on estime que 5 % des patients présentant un aspect de cardiomyopathie hypertrophique (CMH) ont une CA [2] et que 6 à 12 % des patients avec un rétrécissement aortique calcifié (RAC) ont une CA associée au RAC [3].
Les fibres amyloïdes sont constituées d’un assemblage de diverses protéines qui se replient de façon incorrecte avec une organisation particulière riche en feuillets bêta. Elles ont la particularité d’être insolubles. La coloration rouge Congo permet de les mettre en évidence histologiquement, leur conférant une biréfringence caractéristique (vert fluo) en lumière polarisée. Elles peuvent se déposer dans toutes les tuniques cardiaques (myocarde, endocarde, nerf, vaisseaux et valves) [4]. L’atteinte cardiaque peut être isolée ou s’intégrer dans une atteinte systé


Discussion
Aucun commentaire
Commenter cet article