Introduction
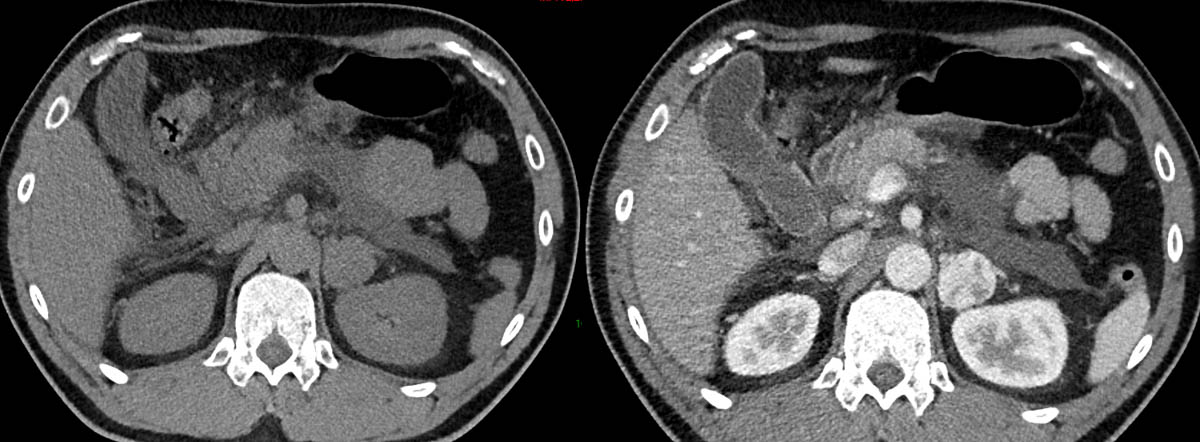
Les phéochromocytomes et les paragangliomes sont des tumeurs neuroendocrines rares, avec une incidence annuelle respective d’environ 4,6 et 1,1 cas par million de personnes [1], et un taux de malignité allant jusqu’à 20 % [2].
Le phéochromocytome (PHEO) est une tumeur de la médullosurrénale qui sécrète des catécholamines (adrénaline, noradrénaline, dopamine). Il représente 3 à 10 % des incidentalomes surrénaliens [3].
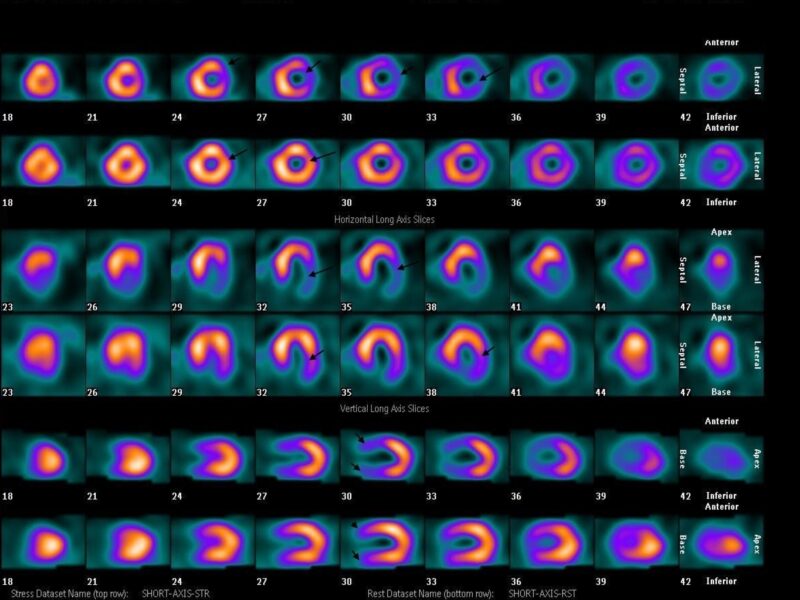
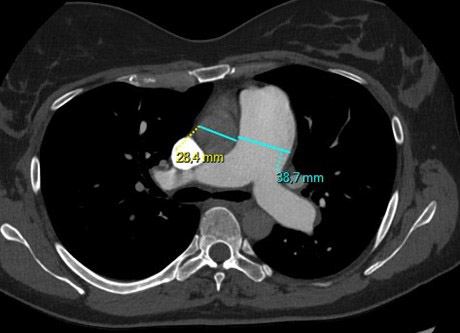
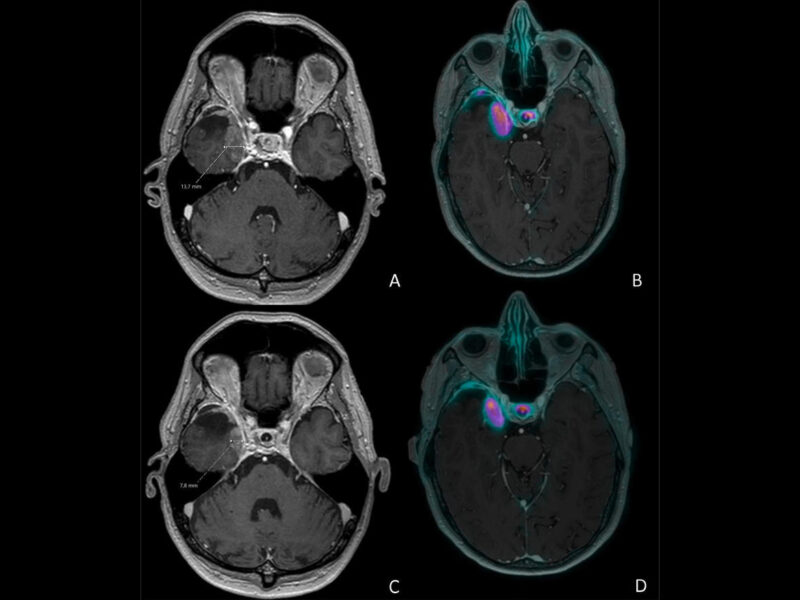
Le paragangliome (PGL) est une tumeur développée en dehors de la surrénale, de la base du crâne au pelvis, aux dépens du tissu chromaffine des ganglions sympathiques pour les lésions médiastinales et abdominales, et du tissu non chromaffine des ganglions parasympathiques pour les lésions de la tête et du cou (3 %). Ces dernières sont en large majorité non sécrétantes (> 99 %) et rarement malignes. 60 % sont localisées au niveau du glomus carotidien [4, 5]. Les PGL médiastinaux et abdominaux sont le plus souvent sécrétants.
La plupart des PGL sont sous-diaphragmat


Discussion
Aucun commentaire
Commenter cet article